| ПОЗНАВАТЕЛЬНОЕ Сила воли ведет к действию, а позитивные действия формируют позитивное отношение Как определить диапазон голоса - ваш вокал Игровые автоматы с быстрым выводом Как самому избавиться от обидчивости Противоречивые взгляды на качества, присущие мужчинам Вкуснейший "Салат из свеклы с чесноком" Натюрморт и его изобразительные возможности Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д. Как научиться брать на себя ответственность Зачем нужны границы в отношениях с детьми? Световозвращающие элементы на детской одежде Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия Классификация ожирения по ИМТ (ВОЗ) Глава 3. Завет мужчины с женщиной
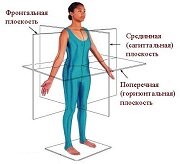
Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
| Утверждены приказом Министра здравоохранения Республики Казахстан от 19 ноября 2009 года № 747
Правила изготовления лекарственных препаратов и изделий медицинского назначения Требования к изготовлению стерильных лекарственных препаратов
В асептических условиях изготавливают:
1) лекарственные препараты для новорожденных;
2) растворы для инъекций и инфузий;
3) ирригационные растворы, вводимые в полости, не содержащие микроорганизмов;
4) жидкие лекарственные препараты для новорожденных и детей до одного года;
5) препараты в виде жидкой лекарственной формы, содержащие антибиотики и другие антимикробные вещества, а также предназначенные для нанесения на раны и ожоговые поверхности;
6) капли глазные, офтальмологические растворы для орошений и примочки;
7) концентрированные растворы (в том числе гомеопатические разведения);
8) жидкие лекарственные препараты в виде внутриаптечной заготовки.
Не допускаются:
1) изготовление стерильных лекарственных препаратов при отсутствии данных о химической совместимости входящих в них лекарственных веществ, технологии и режиме стерилизации, а также при отсутствии методик полного химического контроля;
2) одновременное изготовление на одном рабочем месте нескольких стерильных растворов, содержащих лекарственные вещества с различными наименованиями или одного наименования, но в разных концентрациях.
Результаты контроля отдельных стадий изготовления растворов для инъекций и инфузий регистрируются в журнале регистрации результатов контроля отдельных стадий изготовления растворов для инъекций и инфузий по прилагаемой форме согласно приложению 5 к настоящим Правилам. Журнал должен быть пронумерован, прошнурован, заверен подписью руководителя аптеки и скреплен печатью аптеки.
Контроль стерильных растворов на отсутствие механических включений проводится до и после стерилизации.
Необходимо проверять объем растворов во флаконах (бутылках) и качество их укупорки (металлический колпачок «под обкатку» не должен прокручиваться при проверке вручную, раствор не должен выливаться при опрокидывании флакона (бутылки).
Флаконы с растворами после укупорки маркируются путем надписи, штамповки на крышке или с использованием металлических жетонов с указанием наименования и концентрации.
Стерилизация растворов должна проводиться не позднее трех часов от начала изготовления, под контролем специалиста (фармацевта или провизора).
Не допускается повторная стерилизация растворов.
Регистрация параметров стерилизации производится в журнале регистрации режима стерилизации исходных лекарственных веществ, изготовленных лекарственных препаратов, вспомогательных материалов, посуды по форме согласно приложению 6 к настоящим Правилам. Журнал должен быть пронумерован, прошнурован, заверен подписью руководителя аптеки и скреплен печатью аптеки.
Номенклатура концентратов, полуфабрикатов и внутриаптечной заготовки лекарственных препаратов, изготовляемых в аптеке, ежегодно утверждается органом по сертификации лекарственных средств или аккредитованной испытательной лабораторией, с которой заключен договор о контрольно-аналитическом обслуживании. В данный перечень включают лекарственные препараты, содержащие совместимые активные и вспомогательные вещества, на которые имеются методики анализа для полного химического контроля с установленными сроками годности. Производство стерильных лекарственных средств Принципы
К производству стерильных лекарственных средств предъявляются особые требования, направленные на сведение к минимуму риска загрязнения микроорганизмами, частицами и пирогенами. Выполнение этих требований во многом зависит от опыта персонала, его подготовки и отношения к работе. Особенно высокие требования предъявляются к обеспечению качества, подготовке и выполнению технологических процессов, их тщательной отработке и аттестации (валидации). Контроль конечной стадии производства или контроль готового продукта не может рассматриваться как единственное средство обеспечения стерильности или других показателей качества продукции. Общие положения 1 Производство стерильных препаратов должно быть организовано в чистых помещениях (зонах) с воздушными шлюзами для обеспечения доступа персонала и/или перемещения оборудования и материалов. В чистых помещениях (зонах) должен поддерживаться уровень чистоты по соответствующему стандарту, а воздух должен подаваться через фильтры необходимой эффективности. 2 Подготовка первичной упаковки, приготовление продукта и наполнение должны выполняться в отдельных чистых зонах (помещениях). Процессы производства стерильных лекарственных средств подразделяются на две категории: 3 Чистые помещения (зоны) для производства стерильной продукции классифицируются в соответствии с требованиями к окружающей среде с целью сведения к минимуму риска загрязнения продукта или материалов частицами или микроорганизмами. Каждая производственная операция требует определенного уровня чистоты окружающей среды в эксплуатируемом состоянии. Для обеспечения соответствия чистых помещений (чистых зон) требованиям, предъявляемым к эксплуатируемому состоянию, их проект должен предусматривать достижение заданных классов чистоты воздуха в оснащенной состоянии. Оснащенное состояние - состояние, в котором чистое помещение функционирует, технологическое оборудование полностью укомплектовано, но персонал отсутствует. Эксплуатируемое состояние - состояние чистого помещения, в котором технологическое оборудование функционирует в требуемом режиме с заданным числом работающего персонала. Чистые зоны при производстве стерильных лекарственных средств подразделяются на четыре типа: Примечания (a) Концентрация частиц с размерами, равными или превышающими указанные значения, определяется с помощью дискретного счётчика аэрозольных частиц. В зоне А следует предусматривать непрерывный контроль концентрации частиц. Рекомендуется предусматривать такой контроль и в зоне В*. При текущем контроле зон А и В отбираются пробы общим объёмом не менее 1 м3. Такой же объём пробы рекомендуется и для зоны С*. (b) Уровень загрязнения частицами, показанный в таблице для оснащённого состояния, должен достигаться через 15 - 20 мин (рекомендуемое значение времени восстановления) после завершения процесса при отсутствии персонала. Уровень загрязнения частицами для зоны А в эксплуатируемом состоянии должен поддерживаться в зоне, непосредственно окружающей продукт, когда продукт или открытая упаковка имеют прямой контакт с окружающей средой. Допускается, что в процессе наполнения не всегда может быть показано соответствие стандартам по частицам, поскольку сам продукт может выделять частицы или капельки. (c) В зонах В, С и D кратность воздухообмена должна определяться с учётом размеров помещения, находящегося в нём оборудования и персонала. Для зон А, В и С система подготовки воздуха должна иметь соответствующие фильтры (типа НЕРА). (d) Значения максимально допустимого числа аэрозольных частиц с размерами 0,5 мкм и более в оснащённом и эксплуатируемом состояниях ориентировочно соответствуют классам чистоты по ГОСТ ИСО 14644-1. (e) Предполагается, что в воздухе этих зон частицы с размерами 5 мкм и более должны отсутствовать. Поскольку невозможно статистически достоверно доказать полное отсутствие частиц, для этих случаев установлен предел - 1 частица/м3. Выполнение этого условия следует показать при аттестации чистого помещения. (f) Требования к этой зоне и допустимые пределы зависят от характера выполняемых в ней операций.
Требования к другим параметрам (температуре, относительной влажности и др.) зависят от продукта и характера технологических операций. Эти параметры не связаны с классами чистоты. 4 Следует предусматривать контроль чистоты зон различных типов по частицам в эксплуатируемом состоянии. 5 В асептическом производстве необходимо достаточно часто проводить микробиологический контроль с использованием методов седиментации на чашки, отбора проб в объеме воздуха и с поверхностей (например, смывы и контактные пластины). Методы отбора проб, используемые в эксплуатируемом состоянии, не должны вносить помех в защиту зоны. Результаты контроля следует учитывать при рассмотрении документации на серию готовой продукции. После выполнения критических операций следует проводить микробиологический контроль поверхностей и персонала. Следует также проводить дополнительный микробиологический контроль вне технологического процесса, например, после аттестации (валидации) оборудования, выполнения очистки и дезинфекции. Рекомендуемые пределы допустимого микробного загрязнения чистых зон в эксплуатируемом состоянии приведены в таблице.
6 В зависимости от результатов проводимого контроля следует установить пределы предупреждения и действия для показателей загрязнения частицами и микроорганизмами, а также предусмотреть выполнение корректирующих действий в случае превышения этих пределов. Изолирующая технология 7 Применение изолирующей технологии сводит к минимуму влияние человека на технологические зоны. В асептическом производстве это позволяет значительно снизить риск микробного загрязнения продукции из окружающей среды. В изоляторах и передаточных устройствах всех типов должны выполняться установленные требования к качеству воздуха. При этом следует учитывать, в какой степени возможны утечки (повреждения), вызванные особенностями конструкции или материалов изолятора. Передаточные устройства могут быть разными: от конструкций с одинарной или двойной дверью до полностью герметизированных систем, предусматривающих проведение стерилизации. Процесс передачи материалов внутрь и наружу изолятора является одним из наиболее сильных потенциальных источников загрязнений. Пространство внутри изолятора предназначено для проведения операций, представляющих высокий риск для качества продукции. В то же время допускается организация рабочих зон внутри изолятора без однонаправленного (ламинарного) потока воздуха. Требования к чистоте воздуха в среде, окружающей изолятор, зависят от конструкции изолятора и его назначения. Эта среда должна контролироваться, и для асептического производства соответствовать, по крайней мере, зоне D. 8 Изоляторы могут быть введены в эксплуатацию только после завершения аттестации (валидации), которая должна учитывать все критические факторы изолирующей технологии, например, качество воздуха внутри и снаружи изолятора, порядок обработки изолятора, технологию передачи и целостность изолятора. 9 Следует установить порядок текущего контроля, включающий в себя достаточно частое проведение испытаний на наличие утечек в изоляторе и системе «перчатки-рукава». |