| ПОЗНАВАТЕЛЬНОЕ Сила воли ведет к действию, а позитивные действия формируют позитивное отношение Как определить диапазон голоса - ваш вокал Игровые автоматы с быстрым выводом Как самому избавиться от обидчивости Противоречивые взгляды на качества, присущие мужчинам Вкуснейший "Салат из свеклы с чесноком" Натюрморт и его изобразительные возможности Применение, как принимать мумие? Мумие для волос, лица, при переломах, при кровотечении и т.д. Как научиться брать на себя ответственность Зачем нужны границы в отношениях с детьми? Световозвращающие элементы на детской одежде Как победить свой возраст? Восемь уникальных способов, которые помогут достичь долголетия Классификация ожирения по ИМТ (ВОЗ) Глава 3. Завет мужчины с женщиной
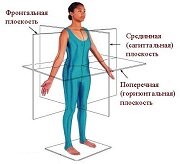
Оси и плоскости тела человека - Тело человека состоит из определенных топографических частей и участков, в которых расположены органы, мышцы, сосуды, нервы и т.д.
| Патогенез наследственных заболеваний.
Проявление генов опосредуется через процессы регуляции белковосинтетических процессов. В цепи ген-признак протекают сложные процессы, зависящие от многих факторов. Одни только структурные гены, непосредственно отвечающие за синтез белка, не в состоянии обеспечить детерминацию развития. В процессе обмена веществ одновременно имеет место активация синтеза не одним, целой группой ферментов, обеспечивающих последовательность определенной цепи реакций, поскольку каждый фермент связан со своим геном структурно-функциональной организации. Согласно процессу генетической регуляции синтеза белка деятельность структурного гена находится под контролем гена-оператора, активность которого, в свою очередь, определяется геном-регулятором, продуктом длительности которого является белок-репрессор, способный связываться с тем или иным веществом, образовавшимся в клетке в процессе обмена. При этом, в зависимости от характера вещества, с которым связывается репрессор, возможно двоякое его действие на оперон: с одной стороны - тормозящее, с другой, если подавляющее влияние репрессора устраняется (связь с веществом) - начинается деятельность соответствующего оперона - активация синтеза. Можно предполагать, что определенные изменения контролирующих генов наряду с мутациями структурных ответственны за возникновение генетически обусловленных болезней. Кроме того, в ряде случаев средовые факторы нарушают реализацию действия нормального гена, т.е. наследственную информацию. Отсюда появляется основание для утверждения, что в ряде случаев заболевания являются связанными не столько с патологией регуляции наследственной информации, сколько с патологией ее реализации. В условиях эксперимента есть возможность заблокировать рецепторное поле клетки - мишень для действия стероидных гормонов с помощью, например, анилиновых красителей. В связи с этим происходит снятие регулирующего влияния гормонов и нарушение синтеза белка - нарушается реализация действия нормального гена. Указанный механизм демонстративен при тестикулярной феминизации - заболевании, при котором формируется псевдогермафродит с наружными гениталиями по женскому типу (внутренние половые органы отсутствуют). При генетическом обследовании выявляется мужской набор половых хромосом, половой хроматин в клетках слизистой отсутствует. Патогенез страдания связан с первичной андрогеноустойчивостью органов-мишеней. Один и тот же мутантный ген у разных организмов может проявить свой эффект различным образом. Фенотипическое проявление гена может варьировать по степени выраженности признака. Это явление связано с экспрессивностью гена - степенью выраженности действия в фенотипическом отношении. Один и тот же признак может проявляться у одних и не проявляется у других особей родственной группы - это явление называется пенетрантностью проявления гена - % особей в популяции, имеющих мутантный фенотип (отношение числа носителей патологического признака к числу носителей мутантного гена). Экспрессивность и пенетрантность характеризуют фенотипические проявления гена, что обусловлено взаимодействием генов в генотипе и различной реакцией генотипа на средовые факторы. Пенетрантность отражает гетерогенность популяции не по основному гену, определяющему конкретный признак, а по модификаторам, создающим генотипическую среду для проявления гена. К модификаторам относят простагландины, активные метаболиты, биоактивные вещества различного происхождения. По характеру изменений генома выделяют следующие мутации: 1. Генные - связанные с одной парой нуклеотидов в полипептидной цепи ДНК (цитологически невидимые изменения). 2. Хромосомные - на уровне отдельной хромосомы (делеция - фрагментация хромосом, приводящая к утрате ее части; дупликация - удвоение участка, перестройки хромосом, обусловленные изменением групп сцепленных генов внутри хромосом - инверсия; перемещение участков - инсерция и др). 3. Геномные - а) полиплоидия - изменение числа хромосом, кратное гаплоидному набору; б) анэуплоидия (гетероплоидия) - некратное гаплоидному набору. По проявлению в гетерозиготе: 1. Доминантные мутации. 2. Рецессивные мутации. По уклонению от нормы: 1. Прямые мутации. 2. Реверсии (часть из них – обратные, супрессорные). В зависимости от причин, вызвавших мутации: 1. Спонтанные 2. Индуцированные По локализации в клетке: 1. Ядерные 2. Цитоплазматические По отношению к особенностям наследования: 1. Генеративные, происходящие в половых клетках 2. Соматические По фенотипу (летальные, морфологические, биохимические, поведенческие, чувствительности к повреждающим агентам и др.). Мутации могут изменить поведение, касаться любых физиологических особенностей организма, вызывать изменение какого-либо фермента и, конечно, затрагивать строение особи. По влиянию на жизнеспособность мутации могут быть летальными и полулетальными, снижающими в большей или меньшей степени жизнеспособность организма. Могут быть практически нейтральными в данных условиях, прямо не влияющими на жизнеспособность и, наконец, хотя и редко, мутации, которые уже при возникновении оказываются полезными. Итак, в связи с этим, согласно фенотипической классификации выделяют: 1. Морфологические мутации, при которых отмечается преимущественно изменение роста и формирования органов. 2. Физиологические мутации - повышающие или понижающие жизнедеятельность организма, полностью или частично тормозящие развитие (полу- и летальные мутации). Существует понятие о летальных генах. Такие гены (обычно в гомозиготном состоянии) или ведут к летальному исходу, или увеличивают его вероятность в раннем эмбриогенезе, или в раннем постнатальном периоде. В большинстве случаев конкретная патология пока не выявлена. 3. Биохимические мутации - мутации, тормозящие или изменяющие синтез определенных химических веществ в организме. Приведенные принципы классификации дают возможность систематизировать наследственные болезни по характеристике генетического дефекта. Классификация форм наследственной патологии. Наследственность и среда играют роль этиологических факторов при любом заболевании, хотя и с разной долей участия. В связи с этим выделяют следующие группы наследственных болезней: 1) собственно наследственные болезни, в которых этиологическую роль играет изменение наследственных структур, роль среды заключается лишь в модификации проявлений заболевания. В эту группу входят моногеннно обусловленные болезни (фенилкетонурия, гемофилия, ахондроплазия), а также хромоомные болезни. 2) экогенетические заболевания, которые также являются наследственными, обусловленными патологическими мутациями, однако для их проявления необходимо специфическое воздействие среды. Например, серповидноклеточная анемия у гетерозиготных носителей при пониженном парциальном давлении кислорода; острая гемолитическая анемия у лиц с мутацией в локусе глюкозо-6-фосфат-дегидрогеназы под влиянием сульфаниламидов. 3) в этой группе многие распространенные заболевания, особенно у пожилых – гипертоническая болезнь, ишемическая болезнь сердца, язвенная болезнь желудка. Этиологическим фактором в их возникновении является средовое воздействие, однако его реализация зависит от индивидуальной генетически детерминируемой предрасположенности организма, в связи с чем эти болезни называют мультифакториальными или болезнями с наследственным предрасположением. С генетической точки зрения наследственные болезни делят на генные и хромосомные. Генные болезни связаны с генными мутациями и далее по количеству затронутых генов выделяют моногенные и полигенные болезни. Выделение моногенных болезней основывается на их сегрегации в поколениях по закону Менделя. Полигенные – болезни с наследственным предрасположением, поскольку предрасположенность является многофакторной. Хромосомные болезни – большая группа патологических состояний, основные проявления которых составляют множественные пороки развития и которые определяются отклонениями в содержании хромосомного материала. Деление наследственных болезней на эти группы не формально. Генные болезни передаются из поколения в поколение без изменений, в то время как большинство хромосомных болезней вообще не передаются, структурные перестройки передаются с дополнительными перекомбинациями. Генные болезни. Ген может мутировать, приводя к изменению или полному отсутствию белка. В связи с этим выделяют отдельные формы генных болезней. Так, нарушение синтеза структурного белка ведет к возникновению пороков развития (синдактилия, полидактилия, брахидактилия, ахондроплазия, микроцефалия и т.д.), нарушение со стороны транспортного белка приводит к функциональным болезням (болезни зрения, слуха и др.), ферментопатии - с нарушением белков – ферментов. По аутосомно-доминантному типу наследуется около 900 болезней: полидактилия, синдактилия и брахидактилия, астигматизм, гемералопия, анонихия, арахнодактилия и ахондроплазия. При аутосомно-рецессивном типе наследования признак проявляется только у особей гомозиготных по данному гену, т.е. когда рецессивный ген получен от каждого родителя. По этому типу наследуется более 800 заболеваний, основная группа – ферментопатии (фенилкетонурия, алкаптонурия, амавротическая идиотия, галактоземия, мукополисахаридозы), различные виды глухоты и немоты. Выделено также и неполное доминирование. Такой тип наследования показан для эссенциальной гиперхолестеринемии: соответствующий ген в гетерозиготном состоянии определяет лишь предрасположенность к гиперхолестеринемии, в гомозиготном же состоянии он приводит к наследственной форме патологии холестеринового обмена – ксантоматозу. Наследование в связи с полом имеет ряд особенностей. Х и Y –хромосомы имеют общие (гомологичные) участки, в которых локализованы гены, наследуемые одинаково как у мужчин, так и у женщин. Например, пигментная ксеродерма, спастическая параплегия, эпидермальный буллез. Негомологичный участок Y-хромосомы (голандрическое наследование) содержит гены перепонок между пальцами и волосатых ушей с передачей только сыновьям. Негомологичный участок Х-хромосомы (рецессивные для женщин и доминантные для мужчин в силу гемизиготности) содержит гены гемофилии, агаммаглобулинемии, несахарного диабета, дальтонизма, ихтиоза. К числу доминантных, полностью сцепленных с полом по Х-хромосоме (с ее негомологичным участком) относятся гипофосфатемический рахит, отсутствие резцов в челюстях. Выявлена также возможность передачи наследственных признаков через цитоплазму яйцеклетки (плазмогены) только через мать – слепота в результате атрофии зрительных нервов (синдром Лебера). Хромосомные болезни отличаются от других наследственных заболеваний тем, что они за редким исключением ограничиваются распространением в пределах одного поколения в связи с полным отсутствием плодовитости у носителей. Тем не менее, хромосомные болезни относятся к группе наследственных, так как они обусловлены мутацией наследственного вещества в половых клетках одного или обоих родителей на хромосомном или геномном уровне. Клинически эти заболевания проявляются тяжелыми нарушениями психики в сочетании с рядом дефектов соматического развития. Хромосомные болезни встречаются в среднем с частотой 1: 250 новорожденных. У 90% эмбрионов с аномалиями хромосом происходит нарушение хромосомного баланса и большая часть прекращает свое развитие на ранних стадиях. Факторы, ведущие к хромосомным аномалиям, по-видимому, общие: 1. Возраст матери. По сравнению со средним возрастом (19-24) у женщин после 35 лет вероятность рождения детей с хромосомными аномалиями возрастает в 10 раз, после 45 лет - в 60 раз. В отношении возраста отцов данных почти нет. Влияние возраста может быть и обратным, например, синдром Шерешевского-Тернера чаще появляется у детей молодых матерей. 2. Ионизирующая радиация - поскольку все виды ионизирующего излучения вызывают хромосомные аберрации в зародышевых и соматических клетках. 3. Вирусные инфекции - корь, краснуха, ветряная оспа, опоясывающий лишай, желтая лихорадка, вирусный гепатит, токсоплазмоз. Хромосомные болезни в своей основе могут иметь либо структурные, либо числовые нарушения как со стороны аутосом, так и хромосом половых клеток. 1. Структурные нарушения аутосом: 5р - утрата короткого плеча (делеция) - синдром «кошачьего крика» - название обусловлено сходством плача ребенка с кошачьем мяуканьем. Это связано с нарушениями ЦНС и с нарушением гортани. Для синдрома характерны также микрогнатия, синдактилия. Отмечается понижение сопротивляемости к инфекциям, поэтому больные погибают рано. Выявляются различные пороки развития (аномалии сердца, почек, грыжи). Встречаются и другие хромосомные аберрации типа делеций: синдромы 4р, 13р, 18р и 18q, 21р, 22q. Транслокации могут быть несбалансированными, что приводит к патологическим состояниям их носителей и сбалансированными - фенотипически не проявляющимися. Структурные нарушения со стороны половых хромосом описаны при синдроме Шерешевского-Тернера со стороны единственной Х-хромосомы (р, q, r, изохромосомы р и q). 2. Числовые нарушения. Аномалии крупных хромосом 1-12 пары обычно летальны. Достаточная жизнеспособность имеет место при трисомии по 21 паре, аномальных половых хромосом и частичных аномалиях. Нуллисомия - отсутствие пары - нежизнеспособность. Моносомия - жизнеспособность только при синдроме ХО. Полиплоидии обычно летальны. Трисомия по 13 паре - синдром Патау - характеризуется множественными пороками головного мозга, сердца, почек, (дети погибают обычно на 3-4 месяце жизни). Трисомия по 18 паре - синдром Эдвардса - множественные дефекты жизненноважных органов, до 1 года обычно доживают не более 7% больных. Транслокационная форма болезни Дауна выражается переносом лишней хромосомы с 22, 4, 15 на 21 пару. Числовые нарушения по половым хромосомам встречаются в виде синдрома Клейнфельтера - ХХУ и его вариантах (ХХХУ, ХХХХУ), характеризуется снижением интеллекта и гипогонадизмом. Известны синдромы ХХХ и варианты, а также ХУУ - в этом случае добавочная У-хромосома влияет больше на поведение, чем на интеллект. Больные агрессивны, отличаются неправильным, даже криминальным поведением. Явление мозаичности связано с разными видами соотношения нормальных и аномальных клеток. В этом случае - промежуточное положение между здоровыми и больными (стертые в клиническом отношении формы). Важным методом предупреждения хромосомных болезней является планирование семьи. Так, в частности, идеальным условием считается зачатие в день овуляции. Также, за 1 месяц до зачатия не должно быть воздействия мутагенов (химических – их основной источник производство; физических – рентгеновское облучение в диагностических или лечебных целях). Особенно опасны вирусные инфекции и соответственно рекомендуется зачатие только спустя 6 месяцев после инфекции. Важно также повышенное введение витаминов – А, С, Е , фолиевой кислоты, микроэлементов – Са, Мg, Zn. Важна также пренатальная диагностика: проводятся скриннинговые обследования с 16 недели оценка a-фетопротеина, при показаниях также амниоцентез, кариограмма, хориондиагностика. |